Vignette: TumorBoost - Normalization of allelic-specific copy numbers in tumors with matched normals (low-level API)
Author: Henrik Bengtsson
Created on: 2009-12-11
Last updated: 2012-02-03
This vignette illustrates how to TumorBoost normalize the allele B
fractions of a tumor given a matched normal using the low-level
normalizeTumorBoost() method available in the aroma.light package. The
method takes the raw allele B fractions for the tumor (betaT) and the
normal (betaN) as numerical vectors for input, and returns the
normalized allele B fractions for the tumor (betaTN) as another vector.
Optionally, it also accepts genotype calls. If not specified, an
internal naive genotyping method is used. For more details on this
implementation, see help(normalizeTumorBoost).
For details on the TumorBoost method, see
Bengtsson, Neuvial, and Speed (2010).
library("aroma.light")
library("R.utils")
# Load the anonymous TCGA data
pathname <- system.file("data-ex/TumorBoost,fracB,exampleData.Rbin", package="aroma.light")
data <- loadObject(pathname)
str(data)
## 'data.frame': 4000 obs. of 4 variables:
## $ position : int 210799615 191229090 115406041 137656361 150926325
## 182084791 3
## 0853337 151390258 167310970 14568632 ...
## $ betaT : num 0.00328 0.59816 0.04127 0.93768 0.05469 ...
## $ betaN : num 0.0057 0.532 0.044 0.9267 0.0319 ...
## $ genotypeN: num 1 0.5 1 0 1 0 0.5 0 1 0 ...
attachLocally(data)
# Plot allele B fractions along the chromosome
layout(matrix(1:4, ncol=1))
par(mar=c(2.5,4,0.5,1)+0.1)
ylim <- c(-0.05, 1.05)
col <- (data[,"genotypeN"] == 1/2) + 1L
pos <- position / 1e6
# Allele B fractions for the normal
plot(pos, betaN, col=col, ylim=ylim)
# Allele B fractions for the tumor (non-normalized)
plot(pos, betaT, col=col, ylim=ylim)
# TumorBoost normalization with (0, 1/2, 1)-genotype estimates
betaTN <- normalizeTumorBoost(betaT=betaT, betaN=betaN, muN=genotypeN)
str(betaTN)
## atomic [1:4000] 0.9976 0.5707 0.9973 0.011 1.0227 ...
## - attr(*, "modelFit")=List of 3
## ..$ method: chr "normalizeTumorBoost"
## ..$ flavor: chr "v4"
## ..$ delta : num [1:4000] -0.9943 0.0275 -0.956 0.9267 -0.9681 ...
# TumorBoost normalization without genotype estimates
betaTN2 <- normalizeTumorBoost(betaT=betaT, betaN=betaN)
str(betaTN2)
## atomic [1:4000] -0.00242 0.57071 -0.00274 1.01096 0.02274 ...
## - attr(*, "modelFit")=List of 3
## ..$ method: chr "normalizeTumorBoost"
## ..$ flavor: chr "v4"
## ..$ delta : num [1:4000] 0.0057 0.0275 0.044 -0.0733 0.0319 ...
plot(pos, betaTN2, col=col, ylim=ylim)
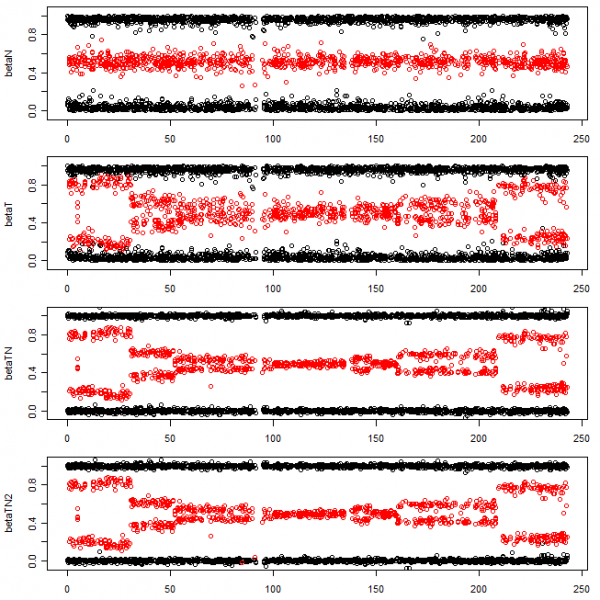
Figure: The allele B fractions for the normal sample (first row), tumor sample (second row), the TumorBoost normalized tumor sample using already estimated genotypes (third row), and the TumorBoost normalized tumor sample without genotypes (fourth row). This is Illumina data on Chromosome 2 from a random TCGA sample (not TCGA-23-1027 used in the TumorBoost manuscript).
References
[1] H. Bengtsson, P. Neuvial, and T. P. Speed. "TumorBoost: normalization of allele-specific tumor copy numbers from a single pair of tumor-normal genotyping microarrays". Eng. In: BMC bioinformatics 11 (May. 2010), p. 245. ISSN: 1471-2105. DOI: 10.1186/1471-2105-11-245. PMID: 20462408.