TumorBoost - Normalization of allelic-specific copy numbers in tumors with matched normals
Author: Pierre Neuvial
Created on: 2010-05-30
Last updated: 2011-03-08
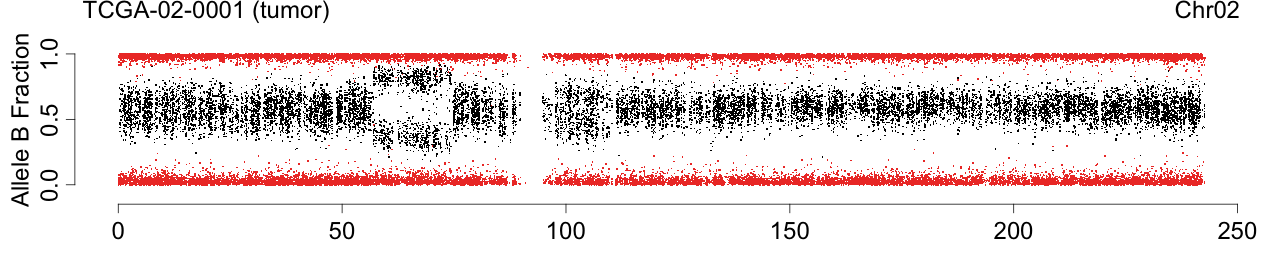
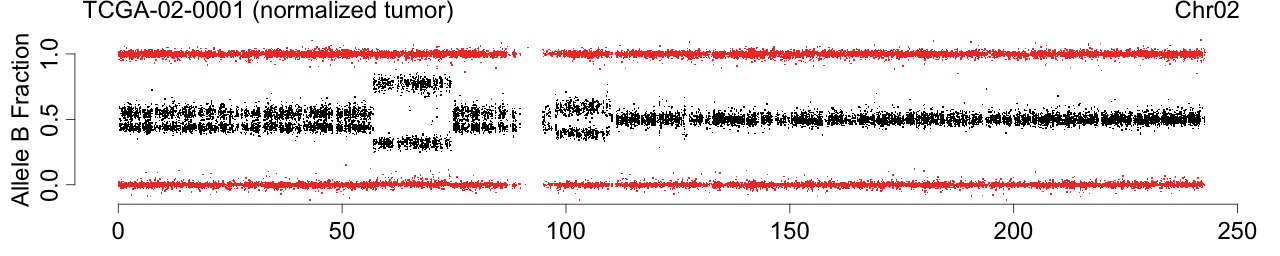
Figure: Allele B fractions before and after TumorBoost normalization.
Introduction
This vignette illustrates the TumorBoost method for normalizing allelic ratios from genotyping microarrays, which is described in Bengtsson, Neuvial, and Speed (2010). The input of the method is a single tumor-normal pair of allelic ratios from genotyping microarrays.
totalAndFracBData/
TCGA,GBM,BeadStudio,XY/
HumanHap550/
TCGA-02-0001-01C-01D-0184-06,fracB.asb
TCGA-02-0001-01C-01D-0184-06,total.asb
TCGA-02-0001-10A-01D-0184-06,fracB.asb
TCGA-02-0001-10A-01D-0184-06,total.asb
TCGA,GBM,CRMAv2/
GenomeWideSNP/
TCGA-02-0001-01C-01D-0182-01,fracB.asb
TCGA-02-0001-01C-01D-0182-01,total.asb
TCGA-02-0001-10A-01D-0182-01,fracB.asb
TCGA-02-0001-10A-01D-0182-01,total.asb
callData/
TCGA,GBM,BeadStudio,XY,NGC/
HumanHap550/
TCGA-02-0001,10A,01D-0184-06,genotypes,confidenceScores.acf
TCGA-02-0001,10A,01D-0184-06,genotypes.acf
annotationData/
chipTypes/
HumanHap550/
HumanHap550,TCGA,HB20080512.ugp
HumanHap550,TCGA,HB20100107,unitNames.txt
GenomeWideSNP_6/
GenomeWideSNP_6,Full.CDF
GenomeWideSNP_6,Full,na26,HB20080821.ugp
Note: In the above data sets, files corresponding to both allelic ratios ('fracB') and total intensities ('total') are listed. Total intensities are only used at the end of this vignette to display total copy number profiles. Total intensities are not used for TumorBoost normalization, and total copy numbers are not affected by TumorBoost normalization.
Input data: genotype calls
To normalize a tumor sample, TumorBoost uses genotype calls (and optionally genotype confidence scores) of the matched normal sample. In this vignette we assume that such genotype calls are available to us (see 'callData' above). In the vignette 'Naive genotype calls and associated confidence scores' we show how such genotype calls can be estimated only based on allelic ratios in the normal sample, as described in the TumorBoost paper.
To use other genotype calls than the naive ones, a data set with similar structure as above has to be created, for example:
callData/
TCGA,GBM,BeadStudio,XY,BeadStudio/
HumanHap550/
TCGA-02-0001,10A,01D-0184-06,genotypes,confidenceScores.acf
TCGA-02-0001,10A,01D-0184-06,genotypes.acf
The creation of such a data set depends on the input data (Birdseed or BeadStudio genotypes for example). It is not documented here.
Input data: allelic ratios
For Affymetrix genotyping microarrays, input data can be obtained using the CRMA v2 method (Bengtsson, Wirapati, and Speed, 2009), e.g.
ds <- doASCRMAv2("TCGA,GBM", chipType="GenomeWideSNP_6,Full")
See CRMA v2 block and CRMA v2 vignette for details.
Setup
library("aroma.cn")
library("R.devices")
devOptions("png", width=1024)
log <- verbose <- Arguments$getVerbose(-8, timestamp=TRUE)
rootPath <- "totalAndFracBData"
rootPath <- Arguments$getReadablePath(rootPath)
dataSets <- c("TCGA,GBM,BeadStudio,XY", "TCGA,GBM,CRMAv2")
dataSet <- dataSets[1] ## Work with Illumina data
# Fullnames translator
fnt <- function(names, ...) {
pattern <- "^(TCGA-[0-9]{2}-[0-9]{4})-([0-9]{2}[A-Z])[-]*(.*)"
gsub(pattern, "\\1,\\2,\\3", names)
} # fnt()
Load the raw (tumor,normal) data set
Here we will assume that matching tumors and normals share the same name but have different tags, e.g. TCGA-02-0001,01C,01D-0184-06 (tumor) and TCGA-02-0001,10A,01D-0184-06 (normal) with the common name TCGA-02-0001.
ds <- AromaUnitFracBCnBinarySet$byName(dataSet, chipType="*", paths=rootPath)
setFullNamesTranslator(ds, fnt)
print(ds)
# Identify all sample names by extracting all unique arrays name (ignoring tags)
sampleNames <- sort(unique(getNames(ds))) # Work with the first sample only
sampleName <- sampleNames[1]
# Extract the two arrays with this name, which should be the tumor and the normal
pair <- indexOf(ds, sampleName)
stopifnot(length(pair) == 2)
# Order as (tumor,normal)
types <- sapply(ds[pair], FUN=function(df) getTags(df)[1])
o <- order(types)
types <- types[o]
pair <- pair[o]
# Extract (tumor, normal) pair
dsPair <- ds[pair]
dsT <- dsPair[1]
print(dsT)
dsN <- dsPair[2]
print(dsN)
This gives:
AromaUnitFracBCnBinarySet:
Name: TCGA
Tags: GBM,BeadStudio,XY
Full name: TCGA,GBM,BeadStudio,XY
Number of files: 1
Names: TCGA-02-0001 [1]
Path (to the first file): totalAndFracBData/TCGA,GBM,BeadStudio,XY/HumanHap550
Total file size: 2.14 MB
RAM: 0.00MB
Load the genotype call set
# Identify available genotype calls
rootPath <- "callData"
rootPath <- Arguments$getReadablePath(rootPath)
genotypeTag <- "NGC"
gsN <- AromaUnitGenotypeCallSet$byName(dataSet, tags=genotypeTag, chipType="*")
setFullNamesTranslator(gsN, fnt)
# Keep only normal genotype files (not needed here, but could be needed in other situations)
types <- sapply(gsN, FUN=function(df) getTags(df)[1])
types <- gsub("[A-Z]$", "", types)
keep <- which(is.element(types, c("10", "11")))
gsN <- gsN[keep]
print(gsN)
This gives:
AromaUnitGenotypeCallSet:
Name: TCGA
Tags: GBM,BeadStudio,XY,NGC
Full name: TCGA,GBM,BeadStudio,XY,NGC
Number of files: 1
Names: TCGA-02-0001 [1]
Path (to the first file): callData/TCGA,GBM,BeadStudio,XY,NGC/HumanHap550
Total file size: 1.07 MB
RAM: 0.00MB
Apply TumorBoost normalization
Recall that we in this vignette assume that matching tumors and normals share the same name but have different tags, e.g. TCGA-02-0001,01C,01D-0184-06 (tumor) and TCGA-02-0001,10A,01D-0184-06 (normal) with the common name TCGA-02-0001. In what follows, we utilize this fact by sorting three data sets such that they are ordered in the same way (as the normal set).
If your tumors and normals do not share the same names this way, you have to make sure the sets are ordered correctly after this step in order for the rest of the analysis to be correct.
# Create a list of matched data sets
dsList <- list(normal=dsN, tumor=dsT, callsN=gsN)
# Make sure they are ordered the same way such that the
# k:th array corresponds to the same sample in all sets.
sampleNames <- getNames(dsList$normal)
dsList <- lapply(dsList, FUN=function(ds) {
idxs <- indexOf(ds, sampleNames)
ds[idxs]
})
print(dsList)
This gives:
$normal
AromaUnitFracBCnBinarySet:
Name: TCGA
Tags: GBM,BeadStudio,XY
Full name: TCGA,GBM,BeadStudio,XY
Number of files: 1
Names: TCGA-02-0001 [1]
Path (to the first file): totalAndFracBData/TCGA,GBM,BeadStudio,XY/HumanHap550
Total file size: 2.14 MB
RAM: 0.00MB
$tumor
AromaUnitFracBCnBinarySet:
Name: TCGA
Tags: GBM,BeadStudio,XY
Full name: TCGA,GBM,BeadStudio,XY
Number of files: 1
Names: TCGA-02-0001 [1]
Path (to the first file): totalAndFracBData/TCGA,GBM,BeadStudio,XY/HumanHap550
Total file size: 2.14 MB
RAM: 0.00MB
$callsN
AromaUnitGenotypeCallSet:
Name: TCGA
Tags: GBM,BeadStudio,XY,NGC
Full name: TCGA,GBM,BeadStudio,XY,NGC
Number of files: 1
Names: TCGA-02-0001 [1]
Path (to the first file): callData/TCGA,GBM,BeadStudio,XY,NGC/HumanHap550
Total file size: 1.07 MB
RAM: 0.00MB
dummy <- lapply(dsList, FUN=function(ds) print(ds[[1]]))
# - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - -
# Normalize allele B fractions for tumors given matched normals
# - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - -
tbn <- TumorBoostNormalization(dsList$tumor, dsList$normal, gcN=dsList$callsN, tags=c("*", "NGC"))
dsTN <- process(tbn, verbose=log)
setFullNamesTranslator(dsTN, fnt)
print(dsTN)
This gives:
AromaUnitFracBCnBinarySet:
Name: TCGA
Tags: GBM,BeadStudio,XY,TBN,NGC
Full name: TCGA,GBM,BeadStudio,XY,TBN,NGC
Number of files: 1
Names: TCGA-02-0001[1]
Path (to the first file): totalAndFracBData/TCGA,GBM,BeadStudio,XY,TBN,NGC/HumanHap550
Total file size: 2.14 MB
RAM: 0.00MB
This has created the following data set:
totalAndFracBData/
TCGA,GBM,BeadStudio,XY,TBN,NGC/
HumanHap550/
TCGA-02-0001-01C-01D-0184-06,fracB.asb
Create a list of matched data sets
dsList <- list(normal=dsN, tumor=dsT, tumorN=dsTN, callsN=gsN)
dsList <- lapply(dsList, FUN=function(ds) {
idxs <- indexOf(ds, getNames(dsList$normal))
ds[idxs]
})
Plots
figPath <- Arguments$getWritablePath("figures")
siteTag <- getTags(ds)
siteTag <- paste(siteTag[-1], collapse=",")
print(siteTag)
## [1] "BeadStudio,XY"
ugp <- getAromaUgpFile(dsList$tumor)
chromosome <- 2
chrTag <- sprintf("Chr%02d", chromosome)
units <- getUnitsOnChromosome(ugp, chromosome=chromosome)
# Identify SNPs only
platform <- getPlatform(ugp)
if (platform == "Affymetrix") {
require("aroma.affymetrix") || throw("Package not loaded: aroma.affymetrix")
snpPattern <- "^SNP"
} else if (platform == "Illumina") {
snpPattern <- "^rs[0-9]"
} else {
throw("Unknown platform: ", platform)
}
unf <- getUnitNamesFile(ugp)
unitNames <- getUnitNames(unf, units=units)
# Identify SNP units
keep <- (regexpr(snpPattern, unitNames) != -1)
units <- units[keep]
pos <- getPositions(ugp, units=units)
# Extract Allele B fractions
kk <- 1
dfList <- lapply(dsList, FUN=`[[`, kk)
beta <- lapply(dfList, FUN=function(df) df[units,1,drop=TRUE])
beta <- as.data.frame(beta)
beta <- as.matrix(beta)
names <- colnames(beta)
names[names == "tumorN"] <- "normalized tumor"
# Plot dimensions
x <- pos/1e6
xlim <- range(x, na.rm=TRUE)
xlab <- "Position (Mb)"
Plot allele B fractions
cols <- as.integer(beta[,"callsN"] != 1) + 1L
for (cc in 1:3) {
toPNG(sampleName, tags=c(siteTag, colnames(beta)[cc], chrTag, "fracB"), aspectRatio=0.6*1/3, {
par(mar=c(1.7,2.5,1.1,1)+0.1, tcl=-0.3, mgp=c(1.4,0.4,0), cex=2)
plot(NA, xlim=xlim, ylim=c(-0.1,1.1), xlab=xlab, ylab="Allele B Fraction", axes=FALSE)
axis(side=1)
axis(side=2, at=c(0,1/2,1))
points(x, beta[,cc], pch=".", col=cols)
label <- sprintf("%s (%s)", sampleName, names[cc])
stext(side=3, pos=0, label)
stext(side=3, pos=1, chrTag)
})
}
Plot total CN
rootPath <- "totalAndFracBData"
rootPath <- Arguments$getReadablePath(rootPath)
dsC <- AromaUnitTotalCnBinarySet$byName(dataSet, chipType="*", paths=rootPath)
setFullNamesTranslator(dsC, fnt)
print(dsC)
This gives:
AromaUnitTotalCnBinarySet:
Name: TCGA
Tags: GBM,BeadStudio,XY
Full name: TCGA,GBM,BeadStudio,XY
Number of files: 2
Names: TCGA-02-0001, TCGA-02-0001 [2]
Path (to the first file): totalAndFracBData/TCGA,GBM,BeadStudio,XY/HumanHap550
Total file size: 4.28 MB
RAM: 0.00MB
pairC <- indexOf(dsC, sampleName)
stopifnot(length(pairC) == 2)
# Order as (tumor,normal)
types <- sapply(dsC[pairC], FUN=function(df) getTags(df)[1])
o <- order(types)
types <- types[o]
pairC <- pairC[o]
# Extract (tumor, normal) pair
dsPairC <- dsC[pairC]
# Extract total CNs
C <- extractMatrix(dsPairC, units=units)
C <- 2*C[,1]/C[,2]
# Plot total CNs
toPNG(sampleName, tags=c(siteTag, chrTag, "CN"), aspectRatio=0.6*1/3, {
par(mar=c(1.7,2.5,1.1,1)+0.1, tcl=-0.3, mgp=c(1.4,0.4,0), cex=2)
plot(NA, xlim=xlim, ylim=c(0,6), xlab=xlab, ylab="Copy number", axes=FALSE)
axis(side=1)
axis(side=2, at=c(0,2,4,6))
points(x, C, pch=".")
label <- sprintf("%s", sampleName)
stext(side=3, pos=0, label)
stext(side=3, pos=1, chrTag)
})
The images generated above are reproduced in Figure 1.
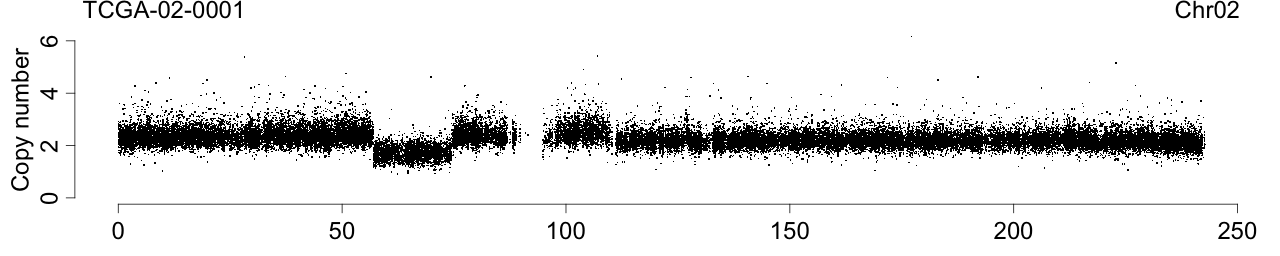
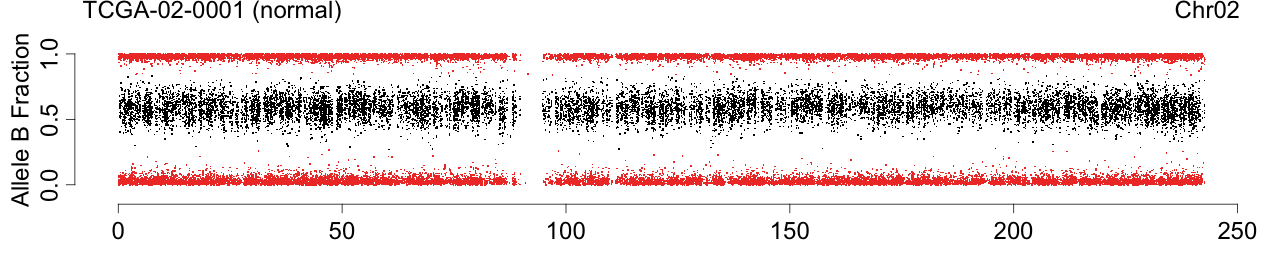
Figure 1. Total copy numbers (top panel), Allele B fractions in the normal (second panel), in the tumor (third panel), and in the normalized tumor (bottom panel). Data are from the Illumina HumanHap550 platform, preprocessed with BeadStudio (method "XY", not "BAF").
References
[1] H. Bengtsson, P. Wirapati, and T. P. Speed. "A single-array preprocessing method for estimating full-resolution raw copy numbers from all Affymetrix genotyping arrays including GenomeWideSNP 5 & 6". Eng. In: Bioinformatics (Oxford, England) 25.17 (Sep. 2009), pp. 2149-56. ISSN: 1367-4811. DOI: 10.1093/bioinformatics/btp371. PMID: 19535535.
[2] H. Bengtsson, P. Neuvial, and T. P. Speed. "TumorBoost: normalization of allele-specific tumor copy numbers from a single pair of tumor-normal genotyping microarrays". Eng. In: BMC bioinformatics 11 (May. 2010), p. 245. ISSN: 1471-2105. DOI: 10.1186/1471-2105-11-245. PMID: 20462408.