Vignette: Empirical probe-signal densities and rank-based quantile normalization
Author: Henrik Bengtsson
Created on: 2009-09-17
Last updated: 2011-02-05
This document illustrated (i) how to create density plots of raw and normalized probe signals and (ii) the result of rank-based quantile normalization stratified by probe type.
Setup
Here we will use 10 public Mapping50K_Hind240 CEL files from the HapMap project.
Raw data
Download the following CEL files from the HapMap site (see the 'HapMap 100K' data set on Page Data Sets):
rawData/
HapMap,CEU,testSet/
Mapping50K_Hind240/
NA06985_Hind_B5_3005533.CEL
NA06991_Hind_B6_3005533.CEL
NA06993_Hind_B4_4000092.CEL
NA06994_Hind_A7_3005533.CEL
NA07000_Hind_A8_3005533.CEL
NA07019_Hind_A12_4000092.CEL
NA07022_Hind_A10_4000092.CEL
NA07029_Hind_A9_4000092.CEL
NA07034_Hind_B1_4000092.CEL
NA07048_Hind_B3_4000092.CEL
Annotation data
Download the following annotation files (Mapping50K_Hind240 & Mapping50K_Xba240):
annotationData/
chipTypes/
Mapping50K_Hind240/
Mapping50K_Hind240.CDF
Mapping50K_Hind240,na26,HB20080916.ufl
Mapping50K_Hind240,na26,HB20080916.ugp
Startup
library("aroma.affymetrix")
library("R.devices")
devOptions("png", width=1024)
# Use a nicer palette of colors
colors <- RColorBrewer::brewer.pal(12, "Paired")
palette(colors)
# Setup the Verbose object
verbose <- Arguments$getVerbose(-10, timestamp=TRUE)
Setup of raw data set
cdf <- AffymetrixCdfFile$byChipType("Mapping50K_Hind240")
print(cdf)
gi <- getGenomeInformation(cdf)
print(gi)
Setup of raw data set
csR <- AffymetrixCelSet$byName("HapMap,CEU,testSet", cdf=cdf)
print(getFullNames(csR))
## [1] "NA06985_Hind_B5_3005533" "NA06991_Hind_B6_3005533"
## [3] "NA06993_Hind_B4_4000092" "NA06994_Hind_A7_3005533"
## [5] "NA07000_Hind_A8_3005533" "NA07019_Hind_A12_4000092"
## [7] "NA07022_Hind_A10_4000092" "NA07029_Hind_A9_4000092"
## [9] "NA07034_Hind_B1_4000092" "NA07048_Hind_B3_4000092"
The CEL files downloaded from HapMap has file names such as NA07000_Hind_A8_3005533.CEL. In order for aroma.affymetrix to identify 'NA07000' as the sample name, and 'A8' and '3005533' as tags (ignore the 'Hind' part), we will utilize so called fullname translators that translates the full name to a comma-separated fullname, e.g. 'NA07000_Hind_A8_3005533' to 'NA07000,A8,3005533'.
setFullNamesTranslator(csR, function(names, ...) {
# Turn into comma-separated tags
names <- gsub("_", ",", names)
# Drop any Hind/Xba tags
names <- gsub(",(Hind|Xba)", "", names)
names
})
print(getFullNames(csR))
## [1] "NA06985,B5,3005533" "NA06991,B6,3005533"
## [3] "NA06993,B4,4000092" "NA06994,A7,3005533"
## [5] "NA07000,A8,3005533" "NA07019,A12,4000092"
## [7] "NA07022,A10,4000092" "NA07029,A9,4000092"
## [9] "NA07034,B1,4000092" "NA07048,B3,4000092"
print(csR)
## AffymetrixCelSet:
## Name: HapMap
## Tags: CEU,testSet
## Path: rawData/HapMap,CEU,testSet/Mapping50K_Hind240
## Platform: Affymetrix
## Chip type: Mapping50K_Hind240
## Number of arrays: 10
## Names: NA06985, NA06991, ..., NA07048
## Time period: 2004-01-14 14:02:08 -- 2004-02-13 11:51:01
## Total file size: 244.78MB
## RAM: 0.01MB
Brief about different types of probes
On Affymetrix arrays, there are different types of probes (cells). The most well known are perfect-match (PM) and mismatch (MM) probes. In addition to these, there are also QC probes, e.g. so called landing-light probes and chequered-flag probes etc.
One can query the CDF for the indices of the cells for each type. If one ask for "all", then all cells on the array are returned.
types <- c("all", "pmmm", "pm", "mm")
cells <- lapply(types, FUN=function(type) identifyCells(cdf, types=type))
names(cells) <- types
str(cells)
## $ all : int [1:2560000] 1 2 3 4 5 6 7 8 9 10 ...
## $ pmmm: int [1:2291560] 1606 1607 1609 1610 1611 1613 1614 1615
## $ pm : int [1:1145780] 1606 1607 1609 1610 1611 1613 1614 1615
## $ mm : int [1:1145780] 3206 3207 3209 3210 3211 3213 3214 3215
As we will see next, one can specify these types also when plotting the empirical densities and when doing quantile normalization.
Raw probe-signal densities
The plotDensity() method for an AffymetrixCelSet estimates and draws
smooth empirical density functions for each array in set. It is possible
to stratify by probe type (see above) by setting the types argument.
For instance, types="all" uses all probes on the chip (regardless of
CDF used), types="pmmm" uses all PM & MM probes, types="pm" only the PM
probes etc. The following code illustrates how to use the types
argument with plotDensity(). See the below figure of the result.
cols <- seq(along=types)
toPNG("plotDensity", tags=c(kk, "raw"), aspectRatio=0.618, {
for (kk in seq(along=types)) {
plotDensity(csR, types=types[kk], col=cols[kk], subset=NULL,
lwd=2, ylim=c(0,0.45), add=(kk > 1))
}
legend("topleft", col=cols, lwd=4, types)
title("Raw probe signals")
})
Rank-based quantile normalization
The QuantileNormalization class provide methods for doing rank-based
quantile normalization of probe signals. By specifying argument
typesToUpdates one can specify what type of probes should be
normalized (and used in the model fitting). All other probe signals are
left unchanged. The default is typesToUpdates="all", but any of "pmmm",
"pm" and "mm" can also be used. The following code illustrates how the
different values produce different results. The results are depicted in
the four panels in the below Figure.
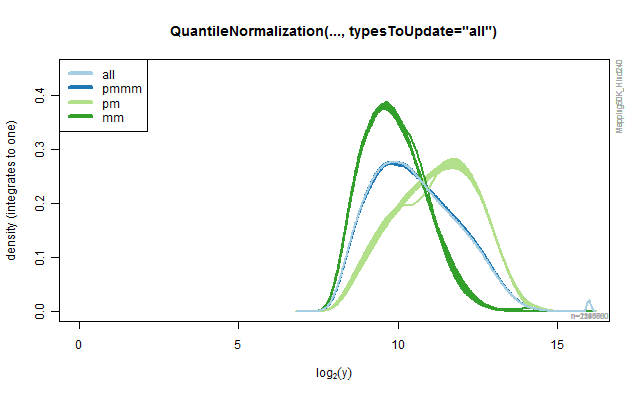
Figure 2A.
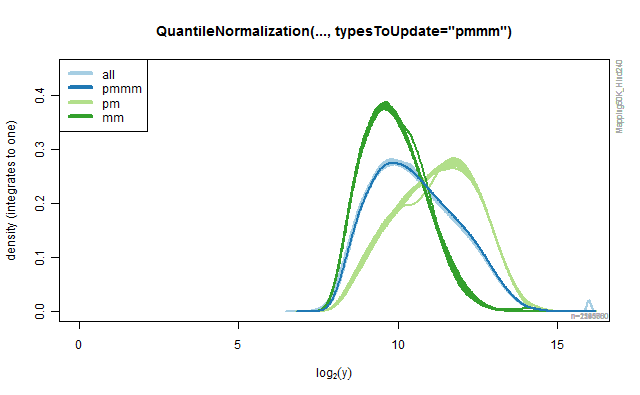
Figure 2B.
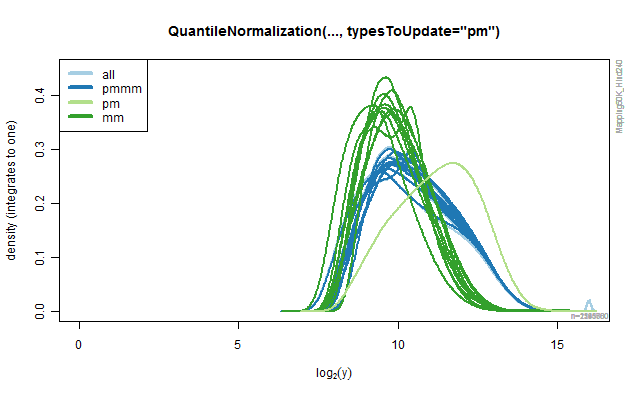
Figure 2C.
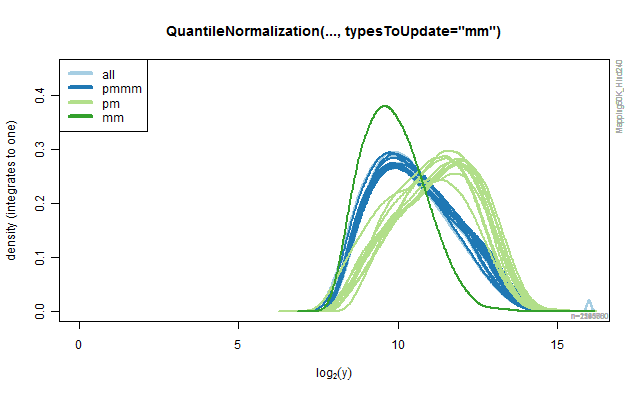
Figure 2D.
for (type in types) {
verbose && enter(verbose, "Rank-based quantile normalization on ", type, " probes")
verbose && cat(verbose, "typesToUpdate: ", type)
qn <- QuantileNormalization(csR, typesToUpdate=type, tags=c("*", type))
print(qn)
csN <- process(qn, verbose=verbose)
toPNG("plotDensity", tags=c("QN", type), aspectRatio=0.618, {
# Plot the type normalized-by last.
kks <- match(c(setdiff(types, type), type), types)
for (kk in kks) {
plotDensity(csN, types=types[kk], subset=NULL, col=cols[kk], lwd=2, ylim=c(0,0.45), add=(kk != kks[1]));
}
legend("topleft", col=cols, lwd=4, types)
title(sprintf("QuantileNormalization(..., typesToUpdate=\"%s\")", type))
})
verbose && exit(verbose)
}